



In the following we list available, currently processed, and finished theses
and student projects. When looking for a topic, please check not only
the available topics but also the processed and recently finished topics. There
might be unannounced but available follow up theses or projects that are not yet
announced.
So if you find a topic interesting, please contact the corresponding supervisor
for further information.
Bioinformatics is a highly specialized application area of computer science and biology
and to successfully solve research questions in this field, you require a lot
of interdisciplinary knowledge.
Therefore, to do a Master thesis with us, we have the minimum
requirement that you have attended one of our
teaching courses.
We may also ask you to present an introductory talk about your chosen topic (given
material provided by us) before we can accept you.
This does not apply to Bachelor theses or projects.
Predicting gene regions in newly sequenced or yet poorly annotated genomes is a vital task in bioinformatics. This way, we can learn about relationships between organisms, related functions of genes and proteins, as well as discover new proteins, with possible implications in medicine and industry. Lately, various deep learning methods (often large language models) have been devised to deliver improved predictions. The goal of this project is to run and compare a selection of these methods, including fine-tuning and possibly additional pre-training of the models. A powerful GPU cluster is available to handle the computations. Extension to thesis possible and also desired. Prerequisites: Python, Linux command line, Git(hub), ideally some practical experience in working with deep learning models (PyTorch) and clusters, plus a keen interest in analysing biological data
Microbiome is the collection of all microbes, such as bacteria, fungi, viruses, along with their genes, which live inside and outside our bodies in all environments surrounding us. To investigate microbiomes, researchers use sequencing data and microbiome analyses. These analyses rely on sequencing data to investigate microbiomes. Such analysis relies on sophisticated computational approaches: assembly, binning, taxonomic classification, functional profiling etc. Analyzing microbiome data makes it possible to answer the two main questions for most microbiome analysis. Who (microorganisms) are there: by extracting the community from the microbiome reads What are they doing (and how): by extracting the gene/pathway abundance profile from the metagenomics reads and transcript abundance profiles from the metatranscriptomics reads and combining them These analyses rely on bioinformatics tools and also databases. Few workflows to process this data are available and most are not openly available, not transparent, or not easy to use by researchers. To tackle this problem, the Freiburg Galaxy team together with the microGalaxy community use Galaxy to build workflows to analyze microbiome sequencing data.
Project context:
MGnify offers an automated pipeline for the analysis and archiving of microbiome data to help determine the taxonomic diversity and functional & metabolic potential of environmental samples.
The pipeline even if documented is not really usable outside their resources. We would like to offer this pipeline for Galaxy users.
This project aims to port the raw reads part of the pipeline into Galaxy.
More information about the project can be found here: https://github.com/usegalaxy-eu/project-ideas/issues/31
The CRISPR-Cas system is an adaptive immune system in many archaea and
bacteria, which provides resistance against invading genetic elements.
The three major components of CRISPR-Cas systems are CRISPR-array,
leader sequence and Cas genes. A recent study[1] demonstrated that there
are proteins adjacent to the Cas proteins that help the CRISPR-Cas to
switch targeting and degrading. This work aims to cluster/classify all
the accessory proteins based on the associated Cas proteins. To do this,
you will use the method from [1] to identify and analysis clusters.
Project Outline
- Start scanning all archaeal and bacterial genomes that have a
CRISPR-Cas system.
- Extract the up-and-downstream flanking genes of each CRISPR-Cas system.
- Classify the genes according to different conditions and find clusters
concerning locations and functions.
[1] https://www.tandfonline.com/doi/full/10.1080/15476286.2018.1483685
You are interested in bleeding edge Linux-Kernel-Technologdy and virtualization?
You want to help to distribute software packages in a OS-independend way?
Than you can help us to solve the deployment problems of scientific software in a general way.
That project will use Docker [1], an open source project that automates the deployment of applications,
to produce self-contained images (containers). These containers are OS independent, versioned
(like a git-history) and easy to use, which enables reproducibility of research results and easy
deployment of entire software stacks.
Prerequisites: Linux/Unix, Bash, autotools
[1] https://www.docker.io
Team-Project: can be combined with the "Graph visualization framework" and the "Galaxy Tool integration" project
Galaxy is an open, web-based platform for data intensive research. The University of Freiburg
is running a Galaxy server to serve all different needs of our researchers. In addition to the
common Next-Gerneration-Sequencing Tools, we offer Tools for cheminformatics [1], proteomics and
RNA bioinformatics. To integrate an apllication into Galaxy, a thin wrapper between the Galaxy
API and the targeted application needs to be written. Here usability is key. Good wrappers are
easy to use and abstracting complicated application details. As part of our Galaxy project we
are permanently seeking for motivated tool-wrappers that are enthusiastic about usability, want
to work with a vibrant community to make Bio- & Cheminformatik Tools accessible for more
researchers. The overall aim is to put the developed wrapper in the Galaxy Tool Shed [2], a Galaxy
Appstore, where everyone can get there favorite application with a few mouse clicks.
Prerequisites: XML, Bash, autotools, Python
[1] https://github.com/bgruening/galaxytools/tree/master/chemicaltoolbox
[2] https://wiki.galaxyproject.org/Tool%20Shed
Team-Project: can be combined with the "Graph visualization framework" and the "Docker" project
The Freiburg Galaxy team is hosting further project ideas in its own GitHub repo.
You want to work on NGS, big-data analysis, Cloud- or HPC-computing or develop
complex front- end backends have a look at the topics in the link below:
https://github.com/bgruening/project-ideas/issues
I'm working on exciting projects to investigate CRISPR-Cas systems and bacteriophages. For the last ten years, CRISPR-Cas systems have become a hot topic, and several problems in the field were already solved using bioinformatics tool. However, there are many exciting aspects of CRISPR-Cas systems that are still open for research. Therefore, we offer many projects for bachelor and master students that are related and attached to our ongoing research (see below for a list of completed projects). The new available projects can be divided into the following categories:
Further Topics are available on request.
If you have a suggestion for a topic you are interested in, do not
hesitate to contact us. Otherwise, the completed theses may lead you.
The aim of this project is to create a low-dimensional representation of SARS-CoV-2 spike protein sequences using an autoencoder neural network. Then, the low dimensional representation of sequences should be clustered using popular clustering algorithms such as TSNE and UMAP to explore if the original differences in sequences belonging to different clades (categories of sequences) are also maintained in lower dimensions. Related reading
SARS-COV2 sequences mutate to multiple variants categorized into lineages and clades, some of which alter the pathogenicity of the virus making it more virulent. Using generative adversarial neural networks, artificial sequences can be generated using the knowledge of the evolution of SARS-COV2 sequences in the past. Ideally, the neural network should learn the 'edit' mechanism of the sequences that evolved in the past and should generate sequences based on the learned knowledge. The generated sequences should be compared with the true sequences to see how good the neural network performs.
For the replication of flaviviruses, the formation of a specific long-range RNA-RNA interaction of the trailing untranslated regions of the virus genomes is crucial. This project aims at the prediction, comparison and modeling of these interactions using state-of-the-art tools for RNA-RNA interaction prediction and RNA alignment to identify common and species-specific details of these interactions.
Microbiome is the collection of all microbes, such as bacteria, fungi, viruses, along with their genes, which live inside and outside our bodies in all environments surrounding us. To investigate microbiomes, researchers use sequencing data and microbiome analyses. These analyses rely on sequencing data to investigate microbiomes. Such analysis relies on sophisticated computational approaches: assembly, binning, taxonomic classification, functional profiling etc. Analyzing microbiome data makes it possible to answer the two main questions for most microbiome analyses. Who (microorganisms) are there: by extracting the community from the microbiome reads What are they doing (and how): by extracting the gene/pathway abundance profile from the metagenomics reads and transcript abundance profiles from the metatranscriptomics reads and combining them. The MGnify Pipeline (https://www.ebi.ac.uk/metagenomics/pipelines/5) provides a standardized way to process metagenomic data and store the results on a public database.
Project context: The MGnify database can be accessed via an API, that allows the retrieval of microbiome abundance data of various origins. Example notebooks that use the API are already included in galaxy. The projects aim to use this abundance data and investigate potential applications in machine learning and comparative metagenomics. Therefore, different normalization approaches need to be applied, that normalize the data in regard to experiment-specific parameters, such as sequencing depth and sample size. The normalized data should be used to investigate the potential to classify samples from different biomes as well as different host phenotypes. The workflows should be implemented and documented in galaxy.
Single-cell multimodal omics allows simultaneous profiling of different types information such as gene expression, DNA methylation, chromatin accessibility and surface protein levels of each individual cells. Such data enables cell characterization based on complex gene regulatory networks. Analysis of such datasets requires immense knowledge in programming languages such as R, python and statistics. To provide experimentalists with complex multimodal analysis workflows, this project aims to integrate computational workflows in Galaxy. We chose to integrate muon based workflows for such data analysis. The muon framework shares datatypes and features with an already Galaxy integrated framework called Scanpy. The objectives of this project are integration of muon multimodal analysis workflows into Galaxy and development of Galaxy training material based on the integrated workflows.
Emerging and powerful technologies like DNA sequencing are getting cheaper and therefore more accessible for many applications, e.g. in microbiome. This produces more data to analyze by scientists. Platforms like Galaxy help scientists to analyze their own (complex) data in a user friendly way. But they need to learn how to do that. The Galaxy Training Network (GTN) created an open-source e-learning infrastructure to provide a collection of tutorials developed and maintained by the worldwide Galaxy community (https://training.galaxyproject.org). Related to microbiome data analysis, the GTN currenlty offers 8 tutorials, built around a research story ( https://training.galaxyproject.org/training-material/topics/metagenomics/). The microGalaxy community aims to expand that catalog for whole-genome microbiome data analysis. The aim is this project is to create a tutorial using data from the Human Microbiome Project, tools an tutorials developed by the Hüttenhover lab to update the general overview tutorial.
Microbiome is the collection of all microbes, such as bacteria, fungi, viruses, along with their genes, which live inside and outside our bodies in all environments surrounding us. To investigate microbiomes, researchers use sequencing data and microbiome analyses. These analyses rely on sequencing data to investigate microbiomes. Such analysis relies on sophisticated computational approaches: assembly, binning, taxonomic classification, functional profiling etc. Analyzing microbiome data makes it possible to answer the two main questions for most microbiome analysis. Who (microorganisms) are there: by extracting the community from the microbiome reads What are they doing (and how): by extracting the gene/pathway abundance profile from the metagenomics reads and transcript abundance profiles from the metatranscriptomics reads and combining them These analyses rely on bioinformatics tools and also databases. Few workflows to process this data are available and most are not openly available, not transparent, or not easy to use by researchers. To tackle this problem, the Freiburg Galaxy team together with the microGalaxy community use Galaxy to build workflows to analyze microbiome sequencing data.
Project context:
MGnify offers an automated pipeline for the analysis and archiving of microbiome data to help determine the taxonomic diversity and functional & metabolic potential of environmental samples.
The pipeline even if documented is not really usable outside their resources. We would like to offer this pipeline for Galaxy users.
This project aims to port the amplicon part of the pipeline into Galaxy.
More information about the project can be found here: https://github.com/usegalaxy-eu/project-ideas/issues/31
Nearly two years after the first report of SARS-CoV-2 in Wuhan, China, the COVID-19 pandemic has affected more than 485 million people. Wastewater surveillance has attracted extensive public attention during the SARS-CoV-2 pandemic, as a passive monitoring system to complement clinical and genomic surveillance activities. Several methods and protocols are already in place that effectively facilitate the detection and quantification of viral RNA in wastewater samples, and concentrations in wastewater have been shown to correlate with trends in reported cases. The Galaxy community has put a lot of efforts for continuous analysis of intra-host variation in SARS-CoV-2 (https://galaxyproject.org/projects/covid19/), including development of workflows. The aim of this thesis are to: (i) Evaluate existing workflows for wastewater data analaysis; (ii) Expand and adaptat existing Galaxy workflows; (iii) Extensive test of workflows on mock and real data; (iv) Connect with existing data sources.
Identifying putative regulatory target regions of bacterial small (s)RNAs is still a challenging problem due to the high false positive rate of predictive methods. One way to greatly reduce false positives is to combine genome-wide predictions of related organisms, which is the core feature of the CopraRNA approach. This project aims at the identification and benchmarking of fast, simple but still sufficiently reliable target prediction workflows based on machine learning techniques to speedup CopraRNA.
Cancer drivers are known as one of the factors which are responsible
for the initialization and progression of cancers.
Therefore, the identification of driver genes plays a crucial point in the development
and improvement of cancer treatment and diagnosis.
Here, we aim at proposing a novel graph neural network-based model to predict cancer drivers
in which various multi-omics data will be taken into consideration.
Prerequisites
Gene prioritization is the task of ordering genes based on their likelihood
of being related to a disease or a set of phenotypes.
Most existing gene prioritization methods are proposed to prioritize genes for diseases.
However, often we observe symptoms and phenotypes and do not know which existing disease
they belong to or they belong to new diseases. Therefore, prioritizing genes directly
from a set of phenotypes is important in practice.
There have been a number of methods proposed in this direction,
but there is still room for improvement.
Here we study to come up with an effective method for ordering genes directly from phenotypes.
Prerequisites
The availability of single-cell RNA sequencing allows us to access
and study the gene expression at a cellular resolution.
Nonetheless, the resulting data are highly noisy since due to amplification and dropout,
causing limited performance for proposing computational methods working on these such data.
In this research, we aim at proposing a graph neural network-based pre-train technique for denoising data.
Our method does not only take gene expression but also the genes' relations encoded
in different data sources into consideration.
Prerequisites
Due to the pandemic situation the interaction with the public face-to-face is not feasible. Therefore, we with the Street Science Community started the development of an online data analysis game (http://streetscience.community/DNAnalyzer/). Within the game users will learn about the microbiome, DNA, sequencing and how to perform a data analysis. Galaxy provides the perfect platform to learn and later perform data analyses. To get scores for there data analysis gamer will share there histories. Within this project a tool will be developed where two shared galaxy history are compared and a score for the submitted history will be calculated. Further information about the topic can be found here: https://github.com/usegalaxy-eu/project-ideas/issues/28
Due to the pandemic situation the interaction with the public face-to-face is not feasible. Therefore the street science community started the development of an online data analysis game (http://streetscience.community/DNAnalyzer/). Within the game users will learn about the microbiome, DNA, sequencing and how to perform a data analysis. Galaxy provides the perfect platform to learn and later perform data analyses. However, the gamer will register on a separate website connected with Galaxy and additionally tracks the successes and results of each gamer. The aim of this project is to implement a small webserver to register participants, display the videos, questions, puzzle, collect and display the score of the participants, and connect with the automated scoring system developed in an other master project. Further information about the topic can be found here: https://github.com/usegalaxy-eu/project-ideas/issues/22
The amount of CPU, RAM, and processing time for a tool to complete is dependent on the size of the input dataset and the complexity of the tool. By emulating these processing requirements with a benchmarking stress-testing tool such as stress-ng, we wish to accurately measure the footprint of the top set of tools on the UseGalaxy.eu workbench with repeated benchmarks, and try to predict their future footprint based on input data size and other extractable content, using machine-learning.
Cancer is a disease that has afflicted the human species for ages, with each tumor possessing its own set of unique characteristics. As a result, people with comparable phenotypes respond to similar therapy in different ways. Largely unsolved, this area has started evolving over the past few decades owing to the availability of multi-omics data and large-scale data of cancer cell lines with different drugs approved for clinical trials. Consequently, a new area termed personalized tumor therapy has emerged. The goal of this research is to propose a novel method that aims at predicting drug response for cancer cell lines.
Studying mRNA expression at single-cell resolution is a well established research area. There exists numerous experimental and computational methods to sequence and analyze the single-cell transcriptomcs. But all of them were designed and optimized to work with protein-coding genes only. Currently, there are only a very few experimental protocols to sequence small non-coding RNAs at single-cell level. It was shown that the existing computational methods that are used for single-cell mRNA-Seq can be used to cluster mature miRNAs and miRNAs also show cell-type specific expressions. In this project we aim to investigate whether miRNAs processing is cell-type specific. To achieve this, we use apply existing computational methods that were developed for bulk miRNA-Seq data to cluster individual cells based on miRNA processing patterns.
The 2020 Nobel Prize in Chemistry to Emmanuelle Charpentier and Jennifer A. Doudna for the discovery and development of CRISPR/Cas9 system highlight the importance of CRISPR-Cas systems. CRISPR-Cas system is an adaptive immune system found in prokaryotic lifeforms and is very diverse in nature. Cas proteins evolve rapidly. Here, we aim to analyse metagenomic data found in the marine ecosystem for the CRISPR-Cas proteins. The main focus is on class-2 type-V system, as the effector protein Cas12 from this system is a promising gene editing candidate. We used three databases for the analysis: Tara Oceans database with 2,631 draft metagenomes, MarRef dataset with 970 assembled metagenome, and IMG/VR dataset with above 90 percent completeness. We built four pipelines comprising different methods and tools for the whole analysis: pipeline 1 for detecting CRISPR-Cas systems and Cas12 proteins, pipeline 2 for transposons, pipeline 3 for repeats and their secondary structures, and pipeline 4 for the spacers and protospacer adjacent motifs (PAMs). We observed that the two tools (CRISPRCasIdentifier and CRISPRCasTyper) used for detecting CRISPR-Cas systems produce very different results, indicating the requirement for building a more accurate and robust tool for the identification of CRISPR-Cas systems. For different variants of Cas12 proteins, we detected different transposable elements. From the analysis of detected repeats, we identified 13 different secondary structures for the repeats found in type V systems and many having a conserved GAAAC or GAA sequence at the 3� terminus. During the spacer analysis, we detected different PAMs. Along with 5� T-rich PAMs, we also detected 5� A-rich PAMs along the upstream of detected spacer sequences. Our work shows that there is still a lot not known about Cas12 proteins, and further in-depth analysis can lead to a better understanding of Cas12 proteins and CRISPR-Cas systems.
In der vorliegenden Bachelorarbeit wird das Verfahren scATAC-Seq und seine biologischen Hintergründe vorgestellt, welches offene Regionen im Chromatin des Genoms einzelner Zellen findet. Des Weiteren wird untersucht, wie die Daten von scATAC-Seq am besten verarbeitet werden, so dass mäglichst viele, hoch qualitative Informationen zu den offenen Chromatinregionen erhalten werden kännen. Dafür werden die Daten speziell vorverarbeitet, anschlie�end werden die Zellen teilweise gruppiert und schlussendlich die Peaks durch Peak Calling bestimmt. Im Anschluss werden die Peaks der einzelnen Zell-Gruppen wieder zusammengefügt, um sie schlussendlich zu vergleichen und auf verschiedene Qualitätskriterien zu überprüfen. In dieser Arbeit werden vier verschiedene Methoden vorgestellt, um diesen Ablauf, mit kleineren Änderungen, durchzuführen. Dazu werden ungefähr 3000, durch scATAC-Seq gewonnene, menschliche Zellen durch die verschiedenen Methoden bearbeitet und untersucht. Anschlie�end werden die Ergebnisse verglichen. Die Resultate zeigen Potential zur Feststellung von diesen Arten der Verarbeitung der Daten. Dabei kann in dieser Arbeit aber nicht eine Methode klar empfohlen werden, da es tiefere Untersuchung der gewonnenen Peaks benätigt, um ein abschlie�endes Urteil über die Qualität der Ergebnisse zu erhalten.
What are the different pressures that can shape genomes in evolution? The aim of this thesis is to focus particularly on the case of reductive genome evolution, i.e. the reduction of genome size over time as observed in some marine cyanobacteria. To address this topic, is used silico artificial evolution, a method in which genomes of virtual organisms evolve via computer simulations, and particularly the Aevol model. Several experiments have been conducted to test the effect of several parameters (population size, mutation rate, and selection strength) on the genome structures and other selection measures (e.g. fitness, robustness).
Drug repurposing is the process of discovering new indications of existing, approved drugs while the latter comprises identifying probable harmful effects of known or novel drugs. It is normally done by in vivo and vitro methods which are of high costs, slow results, and limited sample size besides some ethical issues. Therefore, effective computational methods are needed. In this project, we investigate EHR data and create a machine learning model using the relational graph attention network to predict the potential links between entities of interest link drugs, diagnoses, etc.
Within the 3D space of a cell, DNA forms a structure resembling a ball of wool. The points of contacts of the DNA with itself, called DNA interactions, have "threads" within this "ball of wool" that form smaller loop-like structures called DNA loops. At a close genomic distance, these loops are called topological associated domains (TADs). A z-score based detection algorithm currently exists to detect these TADs, but lacks the ability to detect overlapping TADs and hierarchical structures. In this master project a new approach based on neural networks should be investigated and implemented.
Accurate predictions of off-target sites are key for the safety evaluation of gene
editing using Crispr-cas systems. This thesis investigates factors that involve
the performance of off-target prediction models and proposes an effective model
that is able to overcome the shortcomings of the existing models.
Prerequisites
The goal of this thesis is to assign tissue of action scores to Single Nucleotide Polymorphisms (SNPs)
and genetic loci from Genome Wide Association Studies (GWAS) of kidney function traits using the method published by Torres et al. (Torres et al., 2020).
The method relies on the integration of findings from GWAS with tissue or cell-type specific genomic annotations.
Prerequisites
In collaboration with the Institute of Genetic Epidemiology (University Medical Center),
this project involves the development of an online tool ("scExplorer") to visualize existing scRNA-seq datasets
and to make newly generated scRNA-seq datasets publicly available.
The scExplorer will be used by researchers within a coordinated research initiative to share their scRNA-seq datasets
and make the data accessible for other groups. Researchers will be able to browse different datasets,
display and compare gene expression with different visualization tools for any gene(s) of interest,
as well as to download marker-gene lists for specific cell-types.
The project will be implemented with the R package "shiny".
Prerequisites
While IntaRNA is a state-of-the-art method to predict RNA-RNA interaction, it is not clear if this prediction will happen in nature. We are building a support vector machine model which should validate the in silico interaction on its occurrence in vivo and can therefore be use to post filter interaction predictions. RNA-RNA predictions can experimental be verified by mutation experiments. Based on this experimentally verified interactions we are building we are developing a positive and negative trainings set. This dataset is already discussed in CopomuS by Raden et al.
Current approaches to finding new genes have a high false positive rate. Help us develop a tool to filter candidates in this straightforward Machine Learning project. You will expand on our Scikit-Learn python code and work with state of the art bioinformatics tools. The project covers feature extraction, filtering and classification on an annotated dataset of alignment files.
This project predicts the binding affinities between the potential drugs (ligands) and the target proteins responsible for diseases or conditions.It uses the data of protein-ligand complexes stored in the PDBBind database to train a machine learning model.From every complex, features related to proteins are extracted by using the pocket-finding software fpocket. Four ML models were studied in this project - Simple Linear Regression, Random Forest Regression, Support Vector Regression, and Rotation Forest Regression.
In this thesis, a voxelization procedure was developed and applied to targets (or proteins) in the PCBA (PubChem BioAssay) dataset to create a three-dimensional image of the protein-ligand binding site. These voxelization data were used to train a neural network, more specifically a CNN autoencoder to featurize the binding site by keeping only the most relevant information. This information was then combined with ligand features (which have been calculated using the RDKit descriptor tool from the RDKit library) and finally using machine learning techniques, protein-ligand binding affinity was predicted for each protein-ligand pair.
BioBlend is a Python library to enable simple interaction with Galaxy via the command line or scripts.Galaxy is a data analysis platform for accessible, reproducible and transparent computational research. It includes a web interface through which users can design and perform tasks in a visual and interactive manner. The Galaxy server also exposes this functionality through its REST-based Application Programming Interface (API). In this project several important new features were introduced into BioBlend and the Galaxy API and a tutorial written for future developers.
In recent years, many studies have shown that the three-dimensional conformation of genomes
is a key factor for understanding several important mechanisms on the molecular biological
level. However, the Hi-C experiments typically conducted to measure this 3D-structure are
still expensive, so that computational methods for predicting the spatial chromatin organization
from existing data have recently become subject to research.
In this thesis, two machine learning approaches are investigated with regard to their usability
for predicting chromosome conformation in form of Hi-C contact matrices from ChIP-seq data.
Here, the first method adapts and extends an existing dense neural network architecture for
Hi-C matrix predictions, while the novel second method, Hi-cGAN, leverages techniques from
image synthesis, especially conditional generative adversarial networks (cGANs).
While the dense neural network approach can neither produce satisfactory predictions for the
Hi-C matrices of human cell lines GM12878 and K562, nor for Drosophila Melanogaster embry-
onic cells in the chosen setting, Hi-cGAN yields encouraging outcomes in all three cases.
Within the 3D space of a cell, DNA forms a structure resembling a ball of wool. The points of contacts of the DNA with itself, called DNA interactions, have "threads" within this "ball of wool" that form smaller loop-like structures called DNA loops. At a close genomic distance, these loops are called topological associated domains (TADs). A z-score based detection algorithm currently exists to detect these TADs, but lacks the ability to detect overlapping TADs and hierarchical structures. In this master project a new approach based on machine learning classifiers should be investigated and implemented.
In the 3D space of a cell the DNA forms a structure that looks like a ball of wool. Obviously, many points of contacts of the DNA wire with itself, called DNA interactions, exists in this "ball of wool" and form a structure including DNA loops. These loops contribute to the stability of the DNA and do play an important role in gene regulation. Current research shows that proteins bind on the DNA at these loop locations and contribute to the formation of loops and therefore for the whole structure. The structure of the DNA can be read out with a technique called Hi-C and the resulting data is represented as an interaction matrix in the computer. However, Hi-C is an expensive technique and for many cell types no data is existing while at the same time the technique to read out the position of proteins on the DNA (ChIP-Seq) is quite cheap and a lot of data is online available. The goal of this master project is to use a random forest approach to predict Hi-C interaction matrices by learning the location of proteins. Based on the results of the master project from Andre Bajorat, possible optimizations for this model are investigated.
Within the 3D space of a cell, DNA forms a structure resembling a ball of wool. The points of contacts of the DNA with itself, called DNA interactions, have "threads" within this "ball of wool" that form smaller loop-like structures called DNA loops. At a close genomic distance, these loops are called topological associated domains (TADs). A z-score based detection algorithm currently exists to detect these TADs, but lacks the ability to detect overlapping TADs and hierarchical structures. In this Bachelor thesis a method to detect these was developed and implemented.
Classical linkage analysis is the method of looking for genes that are inherited together in a family tree, which has been now superseded by variant analysis in the era of high-throughput sequencing, but is still relevant in rare disease studies. The Galaxy project is a free and open-source web-based platform for bioinformatic research, and offers users an interactive drag-and-drop avenue to perform their analyses. This project would involve wrapping tools into Galaxy, and chaining them together in a workflow for public user access. Optionally, training material can be written to guide users through the analysis. Applicants need only to know basic HTML/XML and Markdown.
The study of haplotypes is relevant to pedigree analysis, which looks for mutations inherited from founders that manifest only after many generations due to the semi-random/coalescent nature of inheritance. This project will be wrapping an existing haplotype visualization tool into Galaxy, an open source web-based bioinformatic analysis environment, in order to reach a greater number of users. Applicants must know basic Javascript and HTML/XML.
Accessibility-based RNA-RNA interaction prediction methods are typically
modelling a single block of consecutive inter-molecular base pairs.
Thus, interaction pattern that consists of multiple concurrently formed
blocks can not be predicted.
Within this project, we are developing and testing possibilities to
efficiently predict concurrent blocks of interaction within an accessibility-based
prediction model.
The approach will be based on IntaRNA,
which is one of the state-of-the-art programs for RNA-RNA
interaction prediction.
The respective extensions of the IntaRNA package will be integrated into the main
package for external use and further development.
The uncovering of genes linked to human diseases is a pressing challenge in molecular biology and precision medicine. This task is often hindered by a large number of candidate genes and by the heterogeneity of the available information. Therefore, computational methods for the prioritization of candidate genes are needed to deal with these problems. A number of methods have been proposed and have shown potential results. However, there is still a need to develop more accurate disease gene prioritization methods. The aim of this project is to develop a graph neural network-based method for disease gene prioritization. This choice is supported by (1) graphs are a common and natural way to represent the gene relations, and (2) Neural network for graphs are now state-of-the-art in graph (graph node) classification problem.
Triple helix formation has been known to interfere in the gene expression process by often modifying the transcription of targeted genes. Therefore, understanding how and where triple helices form is crucial to better understand gene expression. To identify regions where triple helices formed, wet-lab experiments and some computational methods are performed. However, non-existing methods are based on machine learning. Here we would like to propose a deep learning-based method to detect triple helices in genomic.
CRISPR/Cas9 is a unique and robust gene-editing method that has the ability to accurately edit target genes in a wide variety of organisms. However, experimental results indicate that the binding and cleavage of off-target sequences are a major concern for the application of CRISPR/Cas9 and the sgRNAs should be designed in such a way that the impact of off-targets is minimized. Several computational methods have been proposed as a substitute for expensive lab experiments to predict off-targets. Yet, powerful approaches need to be devised to make precise predictions. Here we aim at proposing a Graph Convolutional Network model to predict off-targets of CRISPR/Cas9. The proposed model is expected to overcome following typical challenges: data imbalance, robustness, prediction crossing different cell-types.
Point mutations are a common way to verify RNA-RNA interactions. So far,
the selection of the position and the introduced mutation is done
manually based on expert knowledge of the experimenter.
Within this project, we are developing and testing possibilities to
automatically evaluate and rank candidate mutations concerning their
potential for interaction validation.
The approach is based on IntaRNA,
which is one of the state-of-the-art programs for RNA-RNA
interaction prediction.
The respective extensions of the IntaRNA package are integrated into the main
package for external use and further development.
The Galaxy-Project, a web platform for big-data biomedical research, needs a lot of computational resources and cloud bursting, e.g. sending excess workloads to the cloud, may be a solution in high-demand situations. But how do the various academic clouds, spread across Europe, perform? May one be better suited than the other for a specific workload? Does physical distance and connectivity between data centers play a big enough role? What about the underlying infrastructure? Do they make a difference, even if the actual instance size is the same? In this work, where I benchmarked various academic clouds in Europe, I want to answer these questions and even offer a framework for future benchmarks, as the need for benchmarking more clouds in the future arise.
Computing base pair probabilities of RNA-RNA interactions allows for a number of useful applications, such as the creation of dot plots, which allow for easy and fast comparison between different base pairing patterns. A number of tools exist that already incorporate base pair probability calculation, such as RNAcofold and NUPACK. However these tools are limited to a specific algorithm for the optimal interaction computation that might lack in precision or computational efficiency depending on the application. IntaRNA on the other hand is a highly exible RNA-RNA interaction prediction tool that implements a large number of different prediction algorithms, including very efficient seed-constraint methods. This thesis explores the benefits and difficulties of introducing the computation of base pair probabilities into a number of IntaRNA predictors, including seed-based predictors. For this reason IntaRNA was extended with the ability to compute base pair probabilities, depending on the chosen prediction model. The output is provided as a dot plot to allow for easy investigation. Finally, a number of applications are presented that bene t from base pair probabilities, including the comparison between verified and non-verified RNA-RNA interactions and the detection of multi-site RNA interactions. Based on these results, potential improvements for IntaRNA's prediction model are discussed, including different approaches for the accessibility computation and the incorporation of sequence conservation into the prediction estimation.
In the 3D space of a cell the DNA forms a structure that looks like a ball of wool. Obviously, many points of contacts of the DNA wire with itself, called DNA interactions, exists in this "ball of wool" and form a structure including DNA loops. These loops contribute to the stability of the DNA and do play an important role in gene regulation. Current research shows that proteins bind on the DNA at these loop locations and contribute to the formation of loops and therefore for the whole structure. The structure of the DNA can be read out with a technique called Hi-C and the resulting data is represented as an interaction matrix in the computer. However, Hi-C is an expensive technique and for many cell types no data is existing while at the same time the technique to read out the position of proteins on the DNA (ChIP-Seq) is quite cheap and a lot of data is online available. The goal of this master project is to use machine learning and neural network regression models/approaches to predict Hi-C interaction matrices by learning the location of proteins.
In the 3D space of a cell the DNA forms a structure that looks like a ball of wool. Obviously, many points of contacts of the DNA wire with itself, called DNA interactions, exists in this "ball of wool" and form a structure including DNA loops. However, many o f these contacts are random contacts or measurement errors and need to be corrected. A Python implementation is existing but comes to its limits for high resolution data caused by high memory usage. This master project should try out if a more efficient algorithm is existing and if an implementation in C++ is possible with less resources.
To evaluate the predicted alignment of the RNA sequence-structure alignment tool LocARNA, so far the alignment score of the has been provided. The score is the optimal value of the objective function from the LocARNA optimization problem. However the scores are not very informative for the end-users, e.g. how well the predicted alignment is significant and likely to occur by chance. It would be desirable to have a statistical measure that not only rank the quality of a given alignment but also make it possible to compare the prediction to other alignment tools and the reference alignment. In this thesis an empirical p-value for LocARNA will be developed. Furthermore, to evaluate a multiple sequence alignment results a suitable scoring schema for multiple sequence alignments will be investigated.
The RNA-RNA interaction prediction tool IntaRNA provides sophisticated and highly accurate results in terms of free energy minimization. Since it is non-trivial for users to interprete the provided free energy terms, this project investigates ways how energy statistics and respective p-values can be provided.
Our group develops the tool
IntaRNA, which is one of the state-of-the-art programs for RNA-RNA
interaction prediction.
We are continously extending the tool (C++, c++11, boost, doxygen, autotools,
openmp) that is hosted on Github BackofenLab/IntaRNA.
This project aims at the implementation and testing of seed-extension
strategies to speedup and improve IntaRNA's predictions.
The developed extensions to IntaRNA will be integrated into the main
package for external use and further development.
Within the last years, we have created interactive implementations of
various algorithms discussed in our lectures. These are freely available
at the
Freiburg RNA tools - Teaching
section of our public webserver.
The algorithms are implemented in Javascript and are accompanied
with according visualizations to better understand each approach.
CRISPR-Cas system of archaea and bacteria provides resistance against viruses and phages. Since phages have a constant battle against prokaryotic; recent discoveries show that have described phage genes that inhibit the CRISPR-Cas function. These are, however, likely to be quite diverse in function as they can interfere with the CRISPR-Cas response at different stages. This work aims to develop a new method of identifying a new family for anti-CRISPR proteins based on homology search.
Archaea and Bacteria are known to acquire immunity against viruses and plasmids through a widely conserved RNA-based gene silencing pathway. This mechanism involves non-coding RNA that originates from Clustered Regularly Interspaced Short Palindromic Repeats, and CRISPR-associated proteins (CRISPR-Cas system). CRISPRs consist of identical repeats that are between 20 to 47 base pairs in length, separated from each other by unique spacer sequences of similar length (27 to 72 base pairs). Most CRISPR arrays are flanked on the upstream (5') side by a leader sequence of 60 to 500 base pairs. These leaders often contain low complexity sequences and are rarely conserved between more distantly related species. Finally, there are the Cas genes, which are usually located directly up- or downstream of CRISPR array, however, they can also be found in very different locations. These genes encode protein complexes which work together with CRISPR arrays to confer the host cell with an adaptive immune system to fight invading viruses and plasmids. This work aims to develop a new tool to detect a CRISPR-Array using machine learning approaches.
Our group develops the tool
MICA, which enables Multiple Interval-based Curve Alignment of arbitrary
curve/profile data. It is currently applied to derive meaningful
consensus data of experimentally measured wood density samples.
Within this project, we will use MICA density profile alignments to
evaluate their potential for crossdating, i.e. the time annotation of
wood samples. Given the increased information compared to standard methods
based on ring widths, the approach should yield high precision even
for small wood samples (few rings).
To benchmark the quality of RNA alignment algorithms, it is important to validate their performance and compare with similar tools. For this purpose a benchmark-pilot framework to automatically benchmark RNA alignment algorithms such as LocARNA and SPARSE will be developed. The aim is to have a modular and easily extendable framework to evaluate various range of tool for different computation platforms from PCs to High Performance Computing grid systems. The task of this project is focused on development of the benchmark-pilot code in python using SnakeMake workflow manager, to replace the previously deployed system.
Our group develops the tool
IntaRNA (see
PhD-thesis A. Richter for details), which is one of the state-of-the-art programs for RNA-RNA
interaction prediction.
We are currently reimplementing and extending the tool (C++, c++11, boost, doxygen, autotools,
openmp) that is hosted on Github BackofenLab/IntaRNA.
This project aims at the implementation and testing of strategies to
enable predictions for very long input molecules, for which the standard
approach might break due to extreme memory consumption. The idea is to
apply a window-based segmentation, which requires a special result
handling to avoid duplications in the output.
The developed extensions to IntaRNA will be integrated into the main
package for external use and further development.
Our group develops the tool
IntaRNA (see
PhD-thesis A. Richter for details), which is one of the state-of-the-art programs for RNA-RNA
interaction prediction.
We are currently reimplementing and extending the tool (C++, c++11, boost, doxygen, autotools,
openmp) that is hosted on Github BackofenLab/IntaRNA.
This project aims at the implementation and testing of new prediction modi, which
incorporate additional constraints to further improve prediction quality.
To this end, an IntaRNA benchmark set and according protocol is compiled
that is used in the course of the thesis to evaluate the newly integrated
features. Furthermore, statistics on known interaction and single-molecule
structures will provide the parameters for the new constraints.
The developed extensions to IntaRNA will be integrated into the main
package for external use and further development.
Within the last years, we have created interactive implementations of
various algorithms discussed in our lectures. These are freely available
at the
Freiburg RNA tools - Teaching
section of our public webserver.
The algorithms are implemented in Javascript and are accompanied
with according visualizations to better understand each approach.
In the course of this project we are focusing on sequence alignment
algorithms as taught in our Bioinformatics-1 and -2 lecture.
Within this project, the state-of-the-art RNA-RNA interaction prediction algorithm implemented in the tool IntaRNA is to be extended. So far, IntaRNA can predict single-site interactions. The project will extend both the underlying theoretical work as well as the IntaRNA implementation to enable the prediction of multi-site interactions. This covers in the first part the extension of the according recursions and their theoretical evaluation. In the second part, the developed theoretical extensions are to be implemented, tuned, and tested. Project requirements
Galaxy is an open, web-based platform for data-intensive research. The University of Freiburg is running a Galaxy server to serve all different needs of our researchers. Visualization is a key aspect in the understanding of data analysis for medical and biological research. The Javascript library BioJS provides powerful visualization of multiple biological data. The overall aim is to integrate specific BioJS modules into Galaxy via its plugin architecture.
Clustering of putative RNAs is currently the major approach for
functional annotation of putative ncRNAs detected in genome-wide
screens. GraphClust is one of the few approaches that can cluster
hundreds of thousands putative ncRNAs as it is based on an
alignment-free approach using an advanced graph kernel. The candidate
clusters are iteratively retrieved and refined using RNA alignment
tools. However the clustering pipeline requires in-depth knowledge
as several tools have to be installed and configured.
The goal of this project is an extension of the GraphClust tool using Galaxy
framework that makes it possible to (a) perform the clustering of
RNAs via a web interface, (b) run the computations on various operating
systems and computation frameworks, (c) freely customize and extend the
generic pipeline for specific needs. The project involves also attempts
to apply the Galaxy workflow on a metatranscriptome dataset.
Ribosome profiling is an emerging technique that with use of deep sequencing methods, gives new insight to translation of proteins from single codon to genome scale. In comparison to former available methods microarrays and RNA-seq, Ribo-seq solely considers active mRNAs at translation phase in a cell which prepare information for protein synthesis. This novel charac- teristic of Ribo-seq provides new data with focus on translation level. The obtained patterns of ribosomal footprints may reveal new aspects in trans- lation field. The aim of this work is to classify Ribo-seq profiles according to different conditions and find clusters with respect to Ribo-seq profiles. This is done by a tool named BlockClust, which is based on a graph kernel method called Neighborhood fast graph kernel (NSPDK). BlockClust en- codes expression profiles data to graphs format and employ NSPDK method for achieving a high performance. Although BlockClust previously applied for clustering non-coding RNAs from their RNA-seq expression profiles, it can also be adapted to use for clustering and classification tasks on other types of data e.g. Ribosome profiling. Therefore, we have adapted Block- Clust by defining new attributes for finding patterns in Ribo-seq data and adding them to the former available set of attributes. Moreover, we per- formed an optimization by using different parameter sets. Furthermore, we showed that it is possible to employ BlockClust on Ribosome profiles. We achieved a good performance in classification of these profiles.
The task of efficiently finding the most similar representatives in a large set of entities is at the core of many problems in a variety of applications, ranging from chemoinformatics to recommendations systems; when the objects of interest are structured entities the problem becomes harder. In these cases structured instances are explicitly converted in sparse vectors that live in very high dimensional spaces (even millions of features). Exact algorithms have unfortunately a computational complexity that scales quadratically with the number of instances times the representation length of each instance, hence these approaches cannot be used when we have a large number of structured instances. A possible solution is to accept approximate results to gain efficiency. The candidate will extended one such approximate technique (the MinHash approximate nearest neighbor scheme) to efficiently solve the neighbor query in sub-linear time. The overall goals of the thesis were to provide an efficient and simple to use implementation for approximate nearest neighbor queries for large collections of high dimensional sparse vectors.
Graph data structures allow us to model complex entities in a natural and expressive way. In the machine learning literature, several types of discriminative systems that can deal with graphs in input are known (e.g. recursive neural networks, graph kernels, graphical models, etc), however, there are few generative approaches that can sample structures belonging to a desired distribution or class. The task of generating samples from a given distribution when this is accessible only via a finite number of examples is well developed when the domain of interest can be embedded in a vector space. The extension of these approaches to structured domains (i.e. where instances are strings, trees, graphs or hyper graphs) is however substantially less developed. One approach for learning constructive systems is based on a variant of the Metropolis Hastings (MH) algorithm guided by an efficient graph grammar, which, crucially, can be efficiently induced from an example set. Such a neighborhood graph based grammar is suitable when the feasibility constraints are local in nature. RNA polymers, which form structures comprising hundreds of nodes (nucleotides), exhibit however dependencies between distant portions of the structure. In order to extend the constructive system to the RNA domain, Mr. Mautner has introduced a multi level strategy based on a notion of graph minors, i.e. graphs obtained by edge contraction operations. An edge contraction is an operation which removes an edge from a graph while simultaneously merging the two vertices that it previously joined. By carefully defining a domain dependent contraction strategy, Mr. Maunter was able to operate on smaller graphs for which local rules are sufficient to capture the feasibility constraints.
A non-coding RNA molecule functionality depends on its structure, which
in turn, is determined by the specific arrangement of its nucleotides. The
inverse folding of an RNA refers to the problem of designing an RNA sequence
which will fold into a desired structure. This is a computationally
complex problem. Algorithms which solve this problem take different
approaches, but they share the following attitude: They start from an initial
sequence or population and try to move it towards a desired product by
performing normal or optimized search methods. RNA inverse folding programs
are given different constraints such as GC-content ranges or basepair
or nucleotide configurations. The output is normally one or more sequences
which fold to the target structure.
This work introduces a basic system that given a set of sample RNA secondary
structures, produces models which generate structures similar to the
sample set. The objectives and constraints are automatically extracted from
samples. For doing this, a system is designed which generates models by
performing learning on families of RNA sequences. This system consists of two
subsystems: one responsible for decomposing secondary structures of sample
RNAs into structural features and building a structural features corpus. It
also extracts neighborhood connectivity models of structural features in the
form of N-grams. The other subsystem is a reinforcement learning framework
which uses the corpus and connectivity rules to produce models for
generating structures which are similar to the samples.
Results in this work show that the current system is able to produce models
from RNA families which have a symmetric shape. To make the system
capable of dealing with a broader range of RNA families and producing
structures with functionalities identical to the sample structures, a refined
feature extraction module has been added to the system. This module extracts
the GC-content, size and local information of structural features and
builds a refined feature corpus. This can provide the basis for a new set of
experiments and a start point for producing models with practical applications.
The energy landscape framework enables the study of the folding
kinetics of molecules. For instance the structure formation process of
single RNA molecules or the interaction formation of two RNAs. To this
end, transition probabilities of one structure to possible successive
structures have to be identified. Unfortunately, there is an exponential
growth of possible structures a molecule can adopt and accordingly an
exponential growth of the energy landscape. One approach to face this
problem is to group structures into "macro-states" and to consider only
transitions between such structure ensembles. But their number is often
still too large to enable kinetics computations.
We have developed an explorative enumeration strategy for such
macro-state partitionings of energy landscapes (see
our article).
Within the Master thesis project of
Bettina Hübner we have investigated and
evaluated different guiding strategies for such an approach
Within this project, the explorative enumeration approach is to be
implemented for RNA energy landscapes.
Project Outline
In recent years many novel RNA species have been discovered by new
sequencing techniques. The correct classification of these RNAs into
new and existing families heavily relies on accurate sequence-structure
alignment tools, which makes it desirable to constantly improve their
alignment quality. Therefore, having a high-performing RNA alignment tool
is of fundamental importance in the field of computational biology.
LocARNA implements an efficient heuristic version of Sankoff's accurate
but computationally expensive algorithm for simultaneous sequence and
structure alignment. The use of heuristics makes the algorithm applicable
in practice, but also forces the inclusion of many additional parameters.
Since the performance of an algorithm depends on the parameter setting,
it is desirable to optimize these settings in order to improve alignment
results. One way to find optimal parameter configurations is to use an
automtic algorithm configuration technique.
In this work the state of the art algorithm configuration tool SMAC is
applied to improve LocARNA 's default parameter settings. The optimization
focuses on fundamental parameters of the LocARNA algorithm. Both global and
local alignment cases are covered, although for the local case this marks the
first in-depth optimization attempt. Hence this work also introduces a complete
local alignment parameter optimization pipeline for LocARNA.
As a result, improved default parameter settings as well as different input
scenario settings for both the global and local alignment cases are proposed.
Notably, the average alignment quality of the local case on an extension of
the Bralibase dataset was improved up to 26%. In conclusion, the presented work
not only managed to optimize LocARNA 's local alignment but also provides a solid
foundation for further works on parameter optimization using the implemented
pipeline.
The CRISPR-Cas system (CRISPR: clustered regularly interspaced short palindromic
repeats, Cas: CRISPR-associated proteins) is an adaptive immune system in bacteria
and archaea, which provides resistance against invading viruses and plasmids. CRISPRs
consist of identical repeats that are between 20 to 40 base pairs in length, separated
from each other by unique spacer sequences of similar length (30 to 50 base pairs).
Most CRISPR arrays are flanked on the upstream (5') side by a leader sequence of
200 to 500 base pairs. These leaders often contain low complexity sequences and are
rarely conserved between more distantly related species. Finally, there are the
cas genes, which are often located directly up- or downstream of CRISPR array,
however, they can also be found in very different locations. These genes encode protein
complexes which work together with CRISPR arrays to confer the host cell with an adaptive
immune system to fight invading viruses and plasmids. Recently, CRISPR-Cas systems were
classified based on their associated Cas proteins. Based on this classification, which
integrates phylogeny, sequence, locus organisation, and content, three types have been
distinguished: Type I, Type II, and Type III
CRISPR-Cas systems.
The classification of Cas proteins has historically been proven difficult because of the
diversity of the proteins involved.
The aim of this work is to cluster/classify all CRISPR-Cas systems based on both associated Cas proteins
and repeat sequence using all available bacteria and archaea genomes. To do this, you will use the in-house
graph kernel approach (EDeN) to identify clusters of related CRISPR-Cas systems and you will compare results
with available classification systems.
Project Outline
Graph data structures allow us to model complex entities in a natural and expressive way. In the machine learning literature, several types of discriminative systems that can deal with graphs in input are known (e.g. recursive neural networks, graph kernels, graphical models, etc), however, there are few generative approaches that can sample structures belonging to a desired distribution or class. The task of generating samples from a given distribution when this is accessible only via a finite number of examples is well developed when the domain of interest can be embedded in a vector space. The extension of these approaches to structured domains (i.e. where instances are strings, trees, graphs or hyper-graphs) is however substantially less developed. While specialized applications exist, e.g. sampling phylogenetic trees, sampling dependency graphs for structural learning in graphical models, or sampling large Web like networks, data driven approaches that can deal with general types of graphs, are still in their infancy. Important applications of a successful generative graph system include the de-novo generation of molecular graphs for drugs and RNA biopolymers with user defined properties derived from prototypical natural examples. In these cases the spatial information of the atom arrangement becomes important for the determination of the associated physicochemical properties. There is therefore the necessity to upgrade these generative graph systems to deal with graphs that can encode spatial information in the form of multiple real valued attributes (e.g. 3D coordinates, distances, angles). In the Thesis the candidate will address the constructive learning problem using a variant of the Metropolis Hastings (MH) algorithm tailored for structural data types. She will upgraded the efficient graph grammar approach of a pre-existing code base to deal with graphs with real valued attributes.
Structured noncoding RNAs perform many functions that are essential for protein synthesis, RNA processing, and gene regulation. Structured RNAs can be detected by comparative genomics, in which homologous sequences are identified and inspected for mutations that conserve RNA secondary structure. To detect novel RNA classes in bacteria and archaea, a variety of bioinformatics strategies have been used, e.g. looking in upstream regions of protein coding genes for cis-regulatory RNAs. To identify ncRNAs independently from protein coding genes, Z. Weinberg has proposed a computational pipeline based on an initial BLAST clustering further refined by looking into secondary structures with CMfinder. The identified structures are then used in homology searches to find homologues that allow CMfinder to further refine its structural alignment. The resulting alignments are scored and then analysed manually to identify the most promising candidates and to infer possible biologic roles.
Synthesis of small molecules that improve on the curative properties of existing drugs or that are effective in previously untreatable illnesses is a very hard task, a task on which pharmaceutical companies are investing enormous amounts of resources. Computational methods become therefore an interesting solution when they can effectively replace the time consuming and expensive design, synthesis and test phases. Since de novo molecule-design systems have to explore a virtually infinite search space, exhaustive searching is infeasible, and they typically resort to local optimisation strategies. To date, one of the most critical aspects is the reliability of the evaluation function invoked to judge the quality of molecules that can be (and generally are) very different from those used in the function induction phase. One possible approach to overcome this difficulty is to integrate the expert knowledge of (medicinal) chemists in the evaluation loop. Doing so in an efficient way is not a trivial task, since one has to 1) minimise the number of times the system resorts to the expensive human oracle, and 2) use a form of interaction suitable for humans.
Bacteria and archaea are known to acquire immunity against viruses and plasmids
through a widely conserved RNA-based gene silencing pathway. This mechanism involves
non-coding RNA that originates from Clustered Regularly Inter-spaced Short Palindromic
Repeats, and CRISPR-associated proteins (CRISPR-Cas system). CRISPRs consist of
identical repeats that are between 24 to 47 base pairs in length, separated from
each other by unique spacer sequences of similar length (27 to 72 base pairs). Most
CRISPR arrays are flanked on the upstream (5') side by a leader sequence of 100 to
500 base pairs. These leaders often contain low complexity sequences and are rarely
conserved between more distantly related species. Finally, there are the Cas genes,
which are oftenlocated directly up- or downstream of CRISPR array, however, they can
also be found in very different locations. These genes encode protein complexes which
work together with CRISPR arrays to confer the host cell with an adaptive immune system
to fight invading viruses and plasmids. The aim of this work is to develop web server
tool for visualizing CRISPR/Cas systems features on/in genome.
Project Outline
Most studies of RNA kinetics use nested structure models to enable at least moderate sequence lengths. Nevertheless, there is evidence that pseudoknot structures are important for the function of some RNA molecules. Thus, ommitting them in kinetics fosters wrong results. This project will compare kinetics based on energy landscape with and without pseudoknot structures. Furthermore, new strategies have to be explored in order to face the vast increase of the landscape size to enable reasonable studies.
The energy landscape framework enables the study of the folding
kinetics of molecules. For instance the structure formation process of
single RNA molecules or the interaction formation of two RNAs. To this
end, transition probabilities of one structure to possible successive
structures have to be identified. Unfortunately, there is an exponential
growth of possible structures a molecule can adopt and accordingly an
exponential growth of the energy landscape. One approach to face this
problem is to group structures into "macro-states" and to consider only
transitions between such structure ensembles. But their number is often
still too large to enable kinetics computations.
We have developed an explorative enumeration strategy for such
macro-state partitionings of energy landscapes (see
our article).
Within the Master thesis project of
Bettina Hübner we have investigated and
evaluated different guiding strategies for such an approach
Within this project, the explorative enumeration approach is to be
implemented for RNA energy landscapes using and extending the already
available algorithms from the
Energy Landscape Library (ELL).
Project Outline
For larger RNA molecules it is often not computationally feasible to enumerate their whole energy landscape. Thus only partial fews of the landscapes are used to compute the kinetics of the respective molecule. Within this project, different strategies are explored to measure the similarity of kinetics, i.e. to evaluate how well the coarse grained model reflects the kinetics based on the complete energy landscape information.
Multiple local alignment of RNA sequences is by now still a challenging problem as parameters for already existing tools are not optimized yet for the local alignment case. The first step to solve this problem is the generation of a local benchmark set to be able to evaluate existing local RNA alignment tools. The main part of this work is the implementation of a pipeline to append genomic context of a given length to an already existing (global) benchmark set. A simple evaluation of LocARNA on the local ncRNA benchmark set and a random test set will be performed.
Small and typically non coding RNAs play a central role in the
regulation of protein biosynthesis in prokaryotic organisms.
Uncovering their regulons in the lab is costly and time intensive.
Recently we developed an algorithm (CopraRNA) to comparatively
predict their direct target mRNAs and consequently their regulons.
We benchmarked the algorithm with one set of organisms (enteric
bacteria). This benchmark yielded results which significantly
outperformed other state of the art algorithms (IntaRNA,
TargetRNA, RNApredator). This project aims at benchmarking CopraRNA
with sets of several differential input data in order to draw
conclusions on which properties the organism dataset should
initially have.
Project Outline
We have developed a new method to to identify the best pairwise alignment
of two curves and an extension to compute a multiple alignment of a set
of curves, namely the
Multiple Interval-based Curve Alignment (MICA).
A prototype of the method was implemented in
R.
Within this project, we want to reimplement and extend the MICA method
in concert with a Graphical User Interface (GUI).
Project Outline
Seven years after the Nobel Prize in Medicine was given to Andrew Fire
and Craig Mello for their initial discovery of RNA interference back in
1993, computational prediction of miRNA target interactions remains a
persistently tough challenge, with little performance improvement over
the past few years. The problem is particularly apparent in animals,
where, opposed to plants, limited complementarity of the miRNA-target
hybrid is sufficient for the interaction to exhibit its regulatory effect.
Ever since the introduction of the first computational prediction
programs, increasingly sophisticated algorithms have tried to tackle this
issue, incorporating more and more predictive features obtained from
observing experimentally verified miRNA target interaction sites. Even
though, as usual, the molecular mechanisms seem to be more diverse and
complicated than expected just a few years ago, there are also
improvements that raise researcher's hopes: Recently, more and more data
sets of several high-throughput methods have become available, allowing
for transcriptome-wide interaction mapping as well as precise localization
of target sites. This in turn enables us to use these data and learn novel
features and modes of miRNA action that can be utilized to build better
performing prediction algorithms.
Project Outline
In this study, we particularly want to use the popular PAR-CLIP data set
that consists of ~ 17000 experimentally observed human miRNA-target
interaction sites to learn novel features from the reported interaction
sites using machine learning techniques. The features of possible interest
consist of sequence and secondary structure information, protein binding
sites, positional and locality data. All features will be integrated into
a graph model to enable a more complete description of the miRNA and mRNA
interaction sites. The overall goal will be to create an algorithmic
pipeline for miRNA-target prediction, whose performance will then be
evaluated and compared to some popular existing prediction algorithms.
To further enhance prediction performance, additional data sets could be
included as training sets in the course of the thesis.
The energy landscape framework enables the study of the folding
kinetics of molecules. For instance the structure formation process of
single RNA molecules or the interaction formation of two RNAs. To this
end, transition probabilities of one structure to possible successive
structures have to be identified. Unfortunately, there is an exponential
growth of possible structures a molecule can adopt and accordingly an
exponential growth of the energy landscape. One approach to face this
problem is to group structures into "macro-states" and to consider only
transitions between such structure ensembles. But their number is often
still too large to enable kinetics computations.
Within this project, different approaches to prune the macro-state
energy landscape represenation are tested in order to reduce the
according transition encoding to a feasible size open for kinetics
computations. The pruning strategies are subject to quantitative and
qualitative evaluations concerning reduced computational requirements
and preserved kinetics quality.
CLIP-seq is a method for genome-wide screening of interactions between RNAs and
RNA-binding proteins. iCLIP is an extension of CLIP-seq that allows locating
RNA-protein interactions with nuceleotide precision. iCLIP employs random
sequence tags in to enable calculation of the number of
binding events from PCR amplified source material. Errors introduced into these
sequence tags during amplification or sequencing can lead to serious overestimation
of binding events. This thesis examins the suitability of RNA barcodes
developed for multiplex sequencing assays to prevent or mitigate this effect.
Chemical molecules can be represented as undirected labeled graphs. The
Neighborhood Substructure Pairwise Distance Kernel (NSPDK) allows for a
thorough description of graph features to enable machine learning based
prediction tasks on such molecules. We
have successfully applied the NSPDK-graph kernel in a first attempt
to predict the aromaticity
property of molecules using trained Support Vector Machines, an important
task in chemoinformatics.
Within this project we want to extensively evaluate the existing approaches
and the potential of our SVM-based aromaticity approach.
Project Outline
The mass flow in a chemical reaction network is determined by the
propagation of atoms from educt to product molecules within each of the
constituent chemical reactions. The Atom Mapping Problem for a given
chemical reaction is the computational task of determining the
correspondences of the atoms between educt and product molecules. We
have introduced a Constraint Programming approach to identify atom mappings
for ``elementary'' reactions (see article).
These feature a cyclic imaginary transition
state (ITS) imposing an additional strong constraint on the bijection
between educt and product atoms. The approach uses Constraint Programming (CP)
techniqus to
identify only chemically feasible ITSs by integrating the cyclic
structure of the chemical transformation into the search.
Within this project, the CP-approach for the identification of chemically
correct atom mappings has to be implemented and extended.
Project Outline
Functional roles of peptide recognition modules (PRMs)
are a key for understanding of various cellular process.
Protein-protein interactions are often mediated by a class of
PRMs such as SH2 and PTB domains. These domains typically bind with
a phosphotyrosine containing peptides to provide a key contribution in
signal transduction. Computational methods that are capable
of modeling the specificity of various PRMs are therefore
of high interest.
Project Outline
The Neighborhood Substructure Pairwise Distance Kernel (NSPDK) allow
us to extract explicit features, occurance count of the all possible pairs of
near small neighbourhood subgroups. The classification would be performed by a
support vector machine (SVM) based on the NSPDK graph kernel.
For making efficient and powerful models that define PRMs specificity, one should consider the high-throughput data.
We are witnessing an increase of the speed at which the result of large
and small scale bioassays is made available in public repositories such
as PubChem through programmatic interfaces.
It is known in literature that activity patterns w.r.t. predefined
bioassays contain valuable information, and can be used to define a
molecular representation in the Activity Space.
Unfortunately the matrix of known compounds vs. experimentally verified
bioassays results is very sparse, as most bioassays provide information
for only few hundreds to thousands compounds. This in turn implies an
incomplete representation in the activity space for most of the compounds.
In order to extend such representation to all compounds we propose to
employ in-silico models to predict the bio-activity for all compounds.
The choice of computational models suitable for this task has to take
into consideration several requirements:
In this thesis the candidate will use a graph kernel (NSPDK) to train a linear max margin model via fast stochastic gradient descent technique. The candidate will set up the necessary infrastructure to perform and monitor the in-silico predictions and develop novel techniques for large scale semi-supervised problems in the chemoinformatics field.
Since the discovery of microRNAs there has been a drive to identify
targets to elucidate their regulatory function. The main features to predict
microRNA targets are the binding of a seed region in the microRNA to the
target, hybridisation energies, and conservation. Computational methods,
however, still predict
hundreds of targets per microRNA and many are incorrect. To improve knowledge
on microRNA function and the accuracy of computational prediction methods,
we need to know more about the targeting mechanism and what features can be
exploited to improve prediction. This knowledge can only be deduced from
experimentally validated interaction sites. Recently, high-throughput methods
have been developed to identify the binding sites of RNA-binding proteins by
combining UV cross-linking of the protein to the RNA, immunoprecipitation,
enzymatic decay, and subsequent sequencing of protected RNA sequences. To
identify microRNA target sites, the method targets Argonaute proteins that
are bound to the microRNA within the RISC complex. Thus, we gain sequences
that are bound to the Argonaute protein in the cell, however, we do not know
which microRNA is involved. Therefore, the respective microRNAs binding to
the identified sequences need to be identified.
Project Outline
The goal of this topic is collect PARCLIP and HITS-CLIP data for mammals and then to develop quality measures to score how likely a microRNA binds to an identified sequence. Furthermore, general properties and statistics of the different datasets are to be explored so that a full understanding of the different characteristics of such data is understood.
RNA-binding proteins (RBPs), which bind to RNA, regulate the translation of RNA and play important roles in post-transcriptional gene expression processes including mRNA splicing, export, stability and mRNA localization. Identifying the binding preferences for a great number of RBPs is not sufficiently achieved, in spite of their important roles in the gene expression. RNA sequences associated with particular RBPs have been favorably characterized by in vitro and in vivo assays; although, particularly designed methods for modeling binding preferences of RBPs are required to deduce an RBP sequence and its structural preferences. Since every method has its own strengths and weaknesses, choosing the right method for a given task is difficult. In this study, an analysis over existing modeling methods is introduced to aid in making the right choice. This analysis consists of several problem sets paired with reference results and a tool to evaluate the results of the estimator methods. Finally, we present the results of applying a benchmark to several methods that includes GraphProt, RNAContext and MatrixREDUCE. This work attempts to find the best way to generate the negative data along with the use of multiple replicates of CLIP-seq method experiments as an advantage to provide training data for applying a benchmark on the modeling methods.
Non-coding RNAs (ncRNAs) form a heterogeneous class of transcripts with little or no protein-coding capacity. Recently, it turned out that these molecules have a plethora of key regulatory roles in eukaryotic cells. NcRNAs directly act at the RNA level without ever being translated to protein. According to their length, one basically distinguishes small (<200bp) and long (>200bp) ncRNAs. The function of a small RNA is typically determined by its secondary structure fold rather than underlying primary sequence. There are several ncRNA classes among small ncRNAs with well defined and well understood secondary structure motifs, examples include micro RNAs (usually forming stem-loop structures) or transfer RNAs (which exhibit the prominent cloverleaf motif). In contrast, it is unclear to which extent long non-coding RNAs contain and are determined by regions of conserved secondary structure.
The aim of this work is to analyse secondary structures of long ncRNAs on a genome-wide scale with state-of-the-art bioinformatic techniques, to possibly identify and further characterise common structural elements shared by these transcripts. This may yield novel insights to the computational de novo prediction of long ncRNAs in recently sequenced eukayotic genomes, one of the open problems in current RNA bioinformatics.
Synthesis of small molecules that improve on the curative properties of existing drugs or that are effective in previously untreatable illnesses is a very hard task, a task on which pharmaceutical companies are investing enormous amounts of resources. On average, development of a new drug takes 10 to 15 years and costs 400-800 million US dollars. Most of the effort though, is spent on investigating compounds that in the end turn out to be unsuitable because of bad ADMET properties. Since only one out of about 5000 screened drug candidates reaches the market, the pharmaceutical industry is looking for ''fail fast, fail cheap'' solutions, i.e. having fast, cheap methods of determining whether the drug candidate does or does not have suitable properties to be a drug and should be rejected. Computational methods become therefore an interesting solution when they can effectively replace the time consuming and expensive design, synthesis and test phases. Amongst such computational methods, those capable to perform de novo molecular design are particularly interesting (Schneider et al., 2005). These approaches produce novel molecular structures with desired pharmacological properties from scratch in an incremental fashion. Since de novo molecule-design systems have to explore a virtually infinite search space2, exhaustive searching is infeasible, and they typically resort to local optimization strategies. Commonly, a de novo design method has to address three questions: how to construct candidate compounds; how to evaluate their potential quality; and how to efficiently sample the search space. To date, one of the most critical aspect is the reliability of the evaluation function. Such function is in fact invoked to judge the quality of molecules that can be (and generally are) very different from those used in the function induction phase therefore leading to unreliable scores. The research project goal is to investigate novel molecular space search strategies. A local optimization algorithm starts with a feasible solution and iteratively tries to obtain a better solution by searching the neighborhood of the current solution. A critical aspect is therefore the choice of the neighborhood structure. Roughly speaking, the larger the neighborhood, the better is the quality of the locally optimal solutions, but the longer it takes to search the neighborhood. In order to define the neighborhood structure current approaches use either the notion of a single vertex/edge, or the notion of molecular fragment. In this latter case expert domain knowledge is used to define meaningful subgraphs (such as functional groups or ring structures) limiting though the search space to the current chemical knowledge available and defined in a machine readable/usable format. Here we want to investigate neighborhood structures for molecular graphs in terms of subgraphs defined implicitly through graph-theoretical notions (that are meaningful in the chemical domain) and not defined explicitly as a pre-specified set of fragments. In particular the thesis will focus on the fast graph kernel models based on the use of neighborhood subgraphs introduced in Costa et al. 2010. Here the subgraphs rooted in each vertex of a molecular graph and induced by all the vertices at bounded small distance from the root have been proved to yield information rich features.
In order to make use of the large amount of genomic information that the sequencing experiments are making available, efficient algorithmic procedures are needed. One of the most fundamental type of processing for genomic data is that of genome alignment, whereby regions belonging to several related genomes are put in bi-univocal correspondence. As a result of the alignment procedure, information of biological relevance can be derived, such as the evolutionary conservation rate of given regions. The sequences in these regions are believed to be important and to correspond to functional biological entities like proteins and non-coding RNA. Correct alignments allow, in other terms, the (semi-)automatic discovery of biological objects (either belonging to known classes, or even to yet unknown classes). However, current genomic alignment techniques 1) are suitable for relatively closely related species, and 2) can process a relatively small number of genomes. In order to allow alignments for thousands of genomes, novel efficient techniques are needed. The choice of computational models suitable for this task has to take into consideration several requirements, such as a) efficiency, b) accuracy and c) flexibility.
Testing to find overlaps between genomic features is an important task in genomics research. We know this feature as intersection. In this project I implement a fast and exible method to find intersections between two sets of genomic intervals by using interval trees. The implementation(unionBed) uses sets of features in BED format as input data and find overlaps between them. Then the unionBed results data is used to analyse three different secondary structure prediction hypotheses for co-transcriptional RNA folding and to compare them to each other.
The prediction of targets of bacterial sRNAs is a very challenging task,
addressed by several approaches. The experimental testing and verification
of sRNA targets is very costly and labour-intensive. Therefore, the
reliable algorithmic prediction of putative sRNA targets could vastly
reduce the amount of wet lab work. However, due to very short and often
imperfect complementarity between the sRNA and its target the prediction
is not a trivial task. The IntaRNA algorithm is one approach, which
frequently, however, does not yield satisfying results yet and therefore
demands improvement.
It has been stated "that it is difficult to make significant target
predictions when searching sequences from a single organism, and that
targets should be predicted in a comparative analysis of multiple
organisms". Eventhough this was stated for eukaryotes, the basic idea of
this thesis also holds for bacteria. The task of improving the IntaRNA
algorithm's prediction quality utilizes exactly this concept, also
incorporating the individual phylogenetic distances between the organisms
analyzed. For instance, there is compelling evidence, that the MicA and
RybB sRNAs in E. coli and Salmonella each have homologous targets in both
organisms, thus indicating a conservation on the regulatory level. Here,
the implementation of the idea that overlapping target predictions for
distinct organisms yield stronger evidence of correct functional
prediction is presented.
The function of ncRNA is determined by its nucleotide composition but also and foremost by its structure, i.e. the identity of paired and unpaired nucleotides. Key analytical tools such as folding, alignment and clustering algorithms, rely on energetic considerations to determin the optimal answer to the specific question they are designed for. These algorithms become inaccurate if the underlying assumptions (energy additivity) they are based upon is violated in reality, i.e. when non-linar effects have to be taken into account. To deal with these problems one can formulate various key questions in terms of non-linar functional dependencies that can be learned from known examples (or parts of examples). Given the importance of the structural element in ncRNA these methods should ideally be able to work in structured domains i.e. should be able to accept in input graph data structures. The methods will belong to the family of kernel machines, since this class of algorithms allows to use heterogeneous features and to accept in input complex instances representation such as sequences or graphs. The aim of the thesis is to develop computational models capable to identify subgraphs within the ncRNA folding graph that are characteristic of biological functions and develop ways to integrate this information into some of the key algorithms for RNA structure analysis.
The goal of the project is to lay the foundations of computing RNA base pair probabilities, as done by the Mc Caskill algorithm, in the framework of Algebraic Dynamic Programming (ADP). In order to concentrate on the essential aspects of this problem, we simplify the scoring model of the algorithm to a Nussinov-style base pair maximization. The main challenge is to compute the outside part, which has no natural correspondence in the grammar parsing framework underlying ADP.
Web frontends of terminal-based bioinformatics tools are important to ease their use for non-computer scientists and to enable ad hoc usage. The project aims at the development of a highly generic web frontend framework generalizing the currently available JSP-based frameworks of the CPSP-web-tools and Freiburg-RNA-tools. Main goals are to simplify the setup of new frontends for arbitrary terminal tools and to develop a robust generic framework. The integration is exemplified by creating a frontend for the recently developed program CARNA.
RNA-alignments are essential for identifying and characterising structured non-coding
RNA. RNA-alignments are different to DNA or protein alignments in the fact that they
not only align according to sequence similarity, but also take the base-pairing patterns
of secondary structures into account. A common procedure to characterise the structure of
non-coding RNAs is to predict the consensus structure of elements of the same family.
The problem with this, is that any errors in the alignment are reflected directly in
the quality of the predicted consensus structure. Therefore, it is of high importance
to get the correct alignment of RNA families. The largest database of such family
alignments is contained in Rfam. A common error in these alignments is that a small
subset has been misaligned with respect to the structure, which results in some
stems slightly offset to either the left or right in comparison to the others.
The goal of this thesis is to develop a method to automatically detect and re-align
these misaligned stems and to thus deliver a quick method to improve these common
errors in the Rfam database. Furthermore, a key part of the work is to understand
the state-of-the art in approaches to align RNA sequences and to perform benchmark
experiments that compare current tools to the here developed method. It is
also important to understand the complexity of measuring the "goodness" of one alignment and
to develop and compare such measures.
There is much evidence in molecular biology that RNA plays an important role
in living cells. Research results in the last decade have shown that protein
coding sequences are only the tip of the iceberg w.r.t. genomic functional
elements. Up to 90% of the genome is transcribed into RNA for which the
function still remains largely unknown.
The structure of an RNA is an important property for its correct function,
e.g. the cloverleaf of a tRNA. However, the experimental determination of
the structure is still a very challenging task, therefore we try to deduce the
structure from the nucleotide sequence, which encodes it.
Furthermore, we find evidence that long RNAs have local regions of functionality
and that the entire sequence does not always contribute to a particular function.
For example cis regulatory elements on mRNA such as SECIS elements and miRNA
binding sites.
In this project we want to analyse long RNA sequences in respect to different
sequence and structure features. The project aims to identify signatures of
natural RNAs and dependencies between RNA sequence and structure.
Sequence features comprise the A,U,G and C
content as well as di-nucleotide and tri-nucleotide content. In terms of
structural features we want to consider accessibilities, base-pair
probabilities, accuracies (MEA) and predictions from tools like RNALfold.
Given these features, we want to identify dependencies between them and between
different sequences.
First, the project involves a graph visualisation for the raw data of single
features and different combinations of features. Because of the huge amount of
data, we need to be able to focus or zoom into regions of interest.
Further, we want to reduce the feature information to only regions of high significance
in comparison to a background model. Thus, a suitable background model needs
to be defined for each feature. With the simplified view, it should be easier to
visually spot correlations between several features at once. After an initial visual
inspection, automatic methods shall be developed to analyse real datasets of different
RNA classes to identify distinct sequence and/or structure signals. First we would
like to concentrate on known cis regulatory elements within the UTRs of mRNAs and
finally we would like to apply the automatic analysis developed
in this thesis to find unknown signals in long non-coding RNAs.
Hundreds of RNA-binding proteins (RBPs) are known. The role of these proteins ranges from mRNA splicing, export, stability and translation regulation. Although RBPs form an important class of proteins, their binding preferences are not well characterized. It is believed that RBPs recognize RNA targets based not only on the sequence information but also on the secondary structure context of the binding. It can be argued that knowledge about RNA secondary structures when searching for the binding motif can be of fundamental importance for certain proteins. The aim of the thesis is to develop computational models capable to identify when the binding preferences of a protein is mainly based on sequence information and when additional structure information is needed. The models will belong to the family of kernel machines, since this class of algorithms allows to use heterogeneous features and to accept in input complex instances representation such as sequences or graphs.
The aim of this work is to help with the implementation and evaluation of the novel algorithm Exparna-P. This algorithm computes all exact pattern matchings between two RNA strands for the entire structure ensemble. In order to speed the algorithm up, a new method needs to be implemented which computes the probability that a position is unpaired under a loop. Then the already existing chaining algorithm has to be slightly modified in order to compute the best set of non-overlapping and non-crossing exact pattern matchings for Exparna-P. The third part of this bachelor thesis is the comparison of the performance of the Exparna-P tool compared to the Exparna tool.
Long ncRNA is a rapidly advancing field of genetics, with yet only briely studied roles (in gene regulation), organization, conservation or medical implications. It is however expected that they will play a great role in further genetic studies and progress. Due to their (sometimes impressive) length (of up to several hundreds of kb) and other particularities, their sequences are rather difficult to align. However, valid sequence alignments are the essential pre-requisite for most subsequent bioinformatic studies of lncRNAs. Therefore, we analyse, compare and benchmark different alignment sets of vertebrate long ncRNAs, namely the Ensembl EPO alignmets, the Galaxy Multiz/TBA blocks and alignments generated by a self-developed pipeline and identify advantages and drawbacks of sequence alignments of lncRNAs.
Despite continued advances in whole genome sequencing techniques and the
development of powerful assembly algorithms, newly sequenced genomes
still often suffer from contaminations during the sequencing process.
The most common sources of contamination are accessory DNAs deliberately
attached to the DNA/RNA under investigation, including vectors,
adapters, linkers, and PCR primers. However, there are also unintended
events, e.g. caused by transposon activity or simply impurities, leading
to contaminated genomic sequences. These may then result in
missassemblies of genomic sequences, meaningless analyses and
potentially erroneous conclusions. However, noone knows to which extent
publicly available genomes are contaminated.
To encompass this unsatisfying situation we therefore plan to develop a
comparative genomics approach to broadly identify contaminations in
available genomic sequences. The project is not only open for
bioinformaticians and computer scientists, it is also suitable for
students with a background in biology.
The number of discovered ncRNAs(non-coding RNAs) that regulate target mRNAs by base pairing is growing fast. This demands for identification of the target mRNAs for those ncRNAs. Thus prediction of such interactions between ncRNAs and mRNAs became of great neccesity to help identify targets for known ncRNAs. A few computational algorithms for this purpose were developed to predict such interactions. While some of the algorithms were fast enough for genome-wide searches, they were not so accurate in predicting interactions between long RNAs. This is because they neglected an important factor for interaction formation which is the interacting site accessibility. IntaRNA considers site accessibility while maintaining the same time and space complexities of these fast algorithms. IntaRNA includes two algorithms, one that gives optimal results according to the Turner free energy model, but is time consuming with time complexity O(n2m2). The second algorithm is heuristic with time complexity O(nm) only, but does not give optimal results for all input sequences. In this thesis we present improvements over both algorithms of IntaRNA. First we modified the non-heuristic algorithm to model more accurately how RNAs are actually forming an interaction. It simulates - in the same order - the sequence of events in which interaction formation is thought to happen in real. The new implementation allows to forbid high energy barriers that might be encountered during interaction formation and that are less likely to be overcome. Second we improved the accuracy of the heuristic algorithm of IntaRNA, making it more accurate and reliable for use in biological researches, without significantly increasing its runtime and space requirements.
At our lab, a general method to align various restricted classes of
pseudoknots has been developed. The alignment scheme has also been
implemented, but due to its generality, it is comparably slow and not
suitable for many large scale practical applications. This work focuses
on developing an efficient implementation of only one specialized
instance of this scheme (The R&G pseudoknot class) that can be used
in real practical scenarios.
The topic is suitable for people interested in algorithms, datastructures, software
development, and C++ programming.
RNA-RNA interaction is a subject of considerable biological relevance as the binding of ncRNA to mRNA can affect both the transcription and translation of the bound mRNA and hence regulate gene expression. The accuracy and reliability of single sequence RNA structure prediction has been shown to increase significantly when the structure of an aligned set of RNA homologs is computed. As such, it is posited that by augmenting an existing RNA-RNA interaction prediction algorithm, that determines an interaction structure based only on thermodynamics, with a phylogenetic component a structure prediction of improved quality can be obtained. This thesis presents the theory, implementation and evaluation of an algorithm that combines thermodynamic and phylogenetic information to predict a consensus interaction structure on a set of aligned mRNAs and ncRNAs.
The Competitive Displacement Detection Method (CDDM) is a method for the
determination of the concentration of an unlabeled target species within
a DNA microarray. This is achieved by introducing fluorescently labeled
oligonucleotides into the microarray that will compete with the target
species for hybridization to the probes. In combination with the
computation of binding kinetics for this competitive displacement
reaction, the measurement of bound competitor can be used to deduce the
concentration of the target.
The goal of this project is the adaptation of this approach to a NASBA
based DNA microarray. Nucleic Acid Sequence Based Amplification (NASBA)
is a method for the amplification of RNA sequences. This amplification
is done within the microarray, obviating the need for external
amplification reactions. With this method the final amount of amplified
product is independent of the initial RNA concentration. Therefore the
determination of target concentrations can only be done indirectly by
considering the progress of the amplification reaction over time. This
adaptation will consist of two main steps:
In this thesis we try to tackle the problem of identifying local RNA elements in a genomewide
scale. We employ a fast sparse algorithm to predict maximum expected accuracy
structures based on base-pairing/unpairing probabilities. Moreover, we introduce a new
locality definition and present an accuracy function reflecting this locality.
Base-pairing and base-unpairing probabilities can be efficiently computed using RNAplfold
included in the Vienna package. Based on these probabilities, we identify structured
regions that have high probabilities of containing significant local RNA motifs.
After that, we introduce our new program RNAMotid together with other included features
that enables it to scan genome-wide sequences for structured regions. Moreover, we
discuss how several modules were integrated together in our program to allow flexibility
and optionality of the analysis.
Finally, we evaluate the performance of RNAMotid in identifying local RNA motifs embedded
in randomly shuffled context. Before that, we apply an overall parameter training
followed by a family-based parameter training. Then we discuss the factors that affect
the performance of RNAMotid.
Messenger (m)RNA is regulated by many different classes of non-coding
(nc)RNAs and proteins to either enhance or inhibit the process of
translation into proteins. The complex mechanisms of how the molecules
identify their specific target sites remains, largely, unknown or a
speculation (e.g for miRNAs).
The aim of this topic is to explore general local structural features
of experimentally verified ncRNA and protein target sites on the
messenger mRNA, i.e. positional entropy, structural complexity, and
accessibility. There already exists evidence of target sites being
more accessible, which means the structure has less base pairs in that
region and is thus "free" for further interaction. A large scale
investigation should give some insight to the influences of these
structural signals within and between various classes of target sites
(e.g. miRNAs, siRNAs, bacterial sRNAs, etc).
The thesis involves the following tasks:
Side chain lattice protein models are a reasonable and necessary extension
of the widely used backbone lattice protein models. To enable folding
simulations a structural neighborhood relation, a so called move set, has
to be defined that is utilizes that enable e.g. Monte-Carlo simulations of
the folding process.
The thesis presents the K-local move set, a local move set defined generically for lattice
protein models. The K-local move set is defined for both backbone and side-chain protein
models via constraint satisfaction problems. The use of the constraint-based approach
enabled its use for an arbitrary lattice. The K-local move set is then used for a simulation
procedure for side-chain protein structures in the face-centered cubic lattice using real
protein sequences and structures.
RNA can be grouped into certain RNA families according structural and functional similarities. Currently, the Rfam 9.1 database (http://rfam.sanger.ac.uk) contains more than 1300 such families. We have already developed a fast index-based (with affix-trees) search method for RNAs. Here, the query is a descriptor and it consists of a stem-loop structure with possible wildcards at different positions. The more sequence information is given the faster is the underlying index-based search engine. On the other hand, if too much sequence information is given, related, but inexact matching stem-loop structure would not be found. Therefore, the goal of this bachelor thesis is to derive such descriptors from Rfam seed-alignments (or other multiple RNA sequence-structure alignments) too feed them into the search engine. If each necessary single descriptor gives a match within a certain region, one could infer a match of the underlying RNA family. A descriptor can been seen therefore as a necessary local motif of an RNA familiy.
The approximate pattern matching problem is the problem of finding all occurences of a certain pattern in a usually much longer text allowing for a fixed error threshold in the matching. The problem has been studied extensively and many very good solutions were found. However, general enough instances of the problem, namely those allowing for generalised error functions, remain with without satisfactory algorithms. This thesis is an attempt to provide such a solution. The new method provided relies on the suffix array data structure to preprocess the text linearly and allow later for fast queries. The new algorithm has the two desirable features of having a fairly simple explanation and implementation and having space and time bounds independent of the size of the alphabet, allowing for arbitrarily large alphabets. Furthermore, the new algorithm handles wildcards quite well while retaining the same time and space worst-case complexities. The algorithms are compared on genuine genetic data from Zebrafish genome and the results are presented. Finally, a parallelized version of the algorithm is presented on CREW-PRAM model of computation. In addition to presenting the new algorithm, several contributions were made to an existing affix array library.
In dieser Masterarbeit präsentieren wir sowohl bekannte, als auch neue Algorithmen zur effzienten Konstruktion und Verwendung von Indexdatenstrukturen. Diese Datenstrukturen haben mannigfaltige Anwendungsmöglichkeiten im Bereich des String-processings. Insbesondere können durch sie Mustersuchen in indexierten Texten beschleunigt werden, wodurch sie eine wichtige Rolle in der Analyse biomolekulare Sequenzen wie z.B. DNA- (Desoxyribonukleinsäure), RNA- (Ribonukleinsäure) und Protein-Sequenzen, spielen.
The combination of the alignment and secondary structure prediction solutions of two RNA sequences can significantly improve the accuracy of the structural predictions. The algorithm which simultaneously solves these problems tends to be computationally expensive like the original form "Sankoff Algorithm" (Sankoff, 1985). Thus, the methods which addressed this problem impose constraints that reduce the computational complexity by restricting the folding and/or alignment and thus make the Sankoff algorithm more practical. In this thesis, reviewing the different Sankoff-style methods in such a way that compares them corresponding to the Sankoff algorithm, through the parallels and differences. As well as, the focus is on the heuristics (i.e. the imposed constraints on the alignments and/or the structures) and comparing between them.
RNAs take part in diverse processes in cells. Energy landscapes can be used to characterize the structural space of an RNA and thus can help us to better understand the processes in which RNAs are involved. The task of estimating energy barriers in RNA landscapes is important in many practical problems such that kinetic RNA folding (Geis et al., 2008) and search for bistable RNA molecules (Flamm et al., 2001). A few approaches has been developed to solve this problem. They need to be improved in two ways: improve time complexity and, at the same time, improve the accuracy of estimations. This master thesis has a task of investigating possible solutions to above-mentioned problem. We apply shape abstraction to the barrier height estimation problem. In the master thesis a number of precise algorithms based on this abstraction have been developed and compared to already existing ones.
There are two conceptually different approaches to predict probable structures of RNA molecules: thermodynamic and kinetic modeling of RNA folding. While purely thermodynamic approaches (e.g. RNAfold) solely consider thermodynamic properties to determine favourable structures, kinetic approaches (e.g. Kinfold) consider the structural changes over a timeframe in addition to their thermodynamic properties. While thermodynamic RNA structure prediction has been extended to RNA-RNA interactions, there doesn't seem to exist a kinetic modeling approach yet. The goal of my diploma thesis will be the implementation and evaluation of two kinetic folding algorithms for RNA-RNA interactions based on stochastic simulations using a Gillespie algorithm as well as macro states.
In dieser Arbeit wurden Signifikanzuntersuchungen für die am Lehrstuhl für
Bioinformatik der Universität Freiburg entwickelten Programme LocARNA und
IntaRNA angestellt. LocARNA bewertet anhand eines
Sequenz-Struktur-Alignments die strukturelle, sowie sequenzielle Ähnlichkeit
zwischen zwei ncRNA Sequenzen, IntaRNA bewertet mögliche Bindestellen
zwischen ncRNA und mRNA, unter Berücksichtigung nicht nur der Sequenz,
sondern auch der Sekundärstruktur der Sequenzen.
Es wurde analysiert, wie sich die ausgegebenen Bewertungen dieser zwei
Programme in Abhängigkeit von den Eigenschaften Länge, AU gegen GC Anteil
und minimaler freier Energie der eingegebenen Sequenzen verändern. Dazu
wurden große Mengen an zufälligen Sequenzen erzeugt, die Verteilung bei
Sequenzen mit gleichen Eigenschaften untersucht und geprüft, wie sich die
Verteilung bei Variation der Länge und des AU zu GC Mengenverhältnisses
ändert. Bei LocARNA wurde mit den Daten eine Support Vektor Maschine
trainiert, die nun für Sequenzpaare die zu erwartende Verteilung angeben
kann. Mit dieser Verteilung als Nullmodell ist es möglich, die P-Werte, und
damit die Signifikanz, der von LocARNA ausgegebenen Bewertungen zu
bestimmen.
Bei IntaRNA wurde festgestellt, dass die ncRNA Sequenzen einen Einfluss auf
die Ausgabe haben, der sich nicht allein durch Länge, AU-Anteil und freier
Energie erklären lässt. Hier sind weitere Untersuchungen nötig, bevor
Gesetzmäßigkeiten bestimmt werden können mit denen die Signifikanz bewertet
werden kann.
Sehr viele Probleme, die sich mit Berechnungen von RNA beschäftigen, die Pseudoknoten
enthalten, sind NP-hart. Am bekanntesten sind hier die Fälle der Strukturvorhersage und
der Berechnung des Alignments. Im Bereich der Strukturvorhersage wurden schon verschiedenste
Algorithmen vorgestellt. Diese beinhalten allerdings Einschränkungen was
die Art der vorkommenden Pseudoknoten angeht, um effizient arbeiten zu können. Diese
hier vorgestellte Arbeit leistet einen Beitrag zu einem neuen Algorithmus, der vom
Lehrstuhl für Bioinformatik vorgestellt wurde. Mit seiner Hilfe ist es möglich, effizient
Sequenz-Struktur-Alignments von RNA Sequenzen zu berechnen, die Pseudoknoten
enthalten. Hierfür wird auf das Prinzip der dynamischen Programmierung zurückgegriffen.
Der Algorithmus besteht dabei im Wesentlichen aus zwei Teilen. Zuerst wird eine der
beiden Sequenzen in einen Parsetree zerlegt und anschliessend das Alignment gebildet.
Das Alignment profitiert hierbei von Einschränkungen auf bestimmte Klassen von Pseudoknoten
auf die selbe Weise wie die jeweilige Strukturvorhersage. Dies hat zur Folge,
dass die Komplexität des Alignments nur um einen linearen Faktor in Bezug auf den
jeweiligen Vorhersagealgorithmus steigt.
Diese Arbeit beschäftigt sich mit dem ersten Teil, dem Berechnen einer Zerlegung der
ersten Sequenz. Hier werden verschiedene Methoden untersucht, wie dies geschehen kann,
sowie diese hinsichtlich ihrer Auswirkungen auf das spätere Alignment analysiert.
ncRNAs are observed to have important roles in transcription, translation and
in post-translation activities. Computational detection of ncRNAs requires sophisticated
methods which take into account structural conservation apart from
sequence information. We present a new pairwise sequence-structure alignment
algorithm, LocARNAp with an aim of obtaining more accurate multiple alignments
than its ancestor LocARNA by using partition function of alignments and
consistency based transformation.
LocARNAp is dynamic programming based sequence-structure alignment algorithm
which computes posterior probabilities of edge alignment from the partition
function of pairwise alignments. Obtained posterior probabilities are then consistency
transformed to include information about other sequences, and thereby
making an improvement in multiple alignment computed using mLocARNA.
We compare the multiple alignments generated by LocARNAp to those obtained
from LocARNA, LARA, STRAL and FoldAlign using benchmark - BRAliBase
2.1�s datasets and extensively study the effect of each parameter setting on the
alignment quality. The analysis of results suggests that our algorithm obtains
overall better quality of results compared to its ancestor - LocARNA and other
algorithms. While there is a huge scope of further improvements, LocARNAp
develops a strong foundation for further research in this direction.
Es werden in der derzeitigen Forschung zwei wesentliche Ansätze für die RNA Faltungsvorhersage verwendet. Zum
einen die direkte Simulation des Faltungsprozesses, bei der über viele Iterationen hinweg
stochastisch Wahrscheinlichkeiten für verschiedene Faltungen/Strukturen bestimmt werden.
Dies ist die am häufigsten genutzte, aber auch bei weitem aufwendigste Methode.
Daher wird oft auf Kinetiken ausgewichen, welche auf einer Vereinfachung der Energielandschaft
basieren. Energielandschaften sind hierbei eine diskrete Beschreibung des Strukturraums
einer RNA, in dem sich der Faltungsprozess abspielt. Zum einen werden ganze
Teile der Energielandschaft zu sogenannten Macrostates zusammengefasst, zum anderen
wird die Landschaft oft vereinfachend durch BarrierTree repräsentiert wodurch Adjazenzinformationen
zugunsten einer effizienten Berechnung verworfen werden.
In dieser Arbeit wird ein Hybridansatz vorgestellt, welcher die häufig verwendeten Macrostate- und Arrheniuskinetiken miteinander
verknüpft. Für den unteren Bereich der Energielandschaft wird die Macrostatekinetik
verwendet, während im oberen Bereich durch ein Sampling der Übergangshöhen eine
Arrheniuskinetik möglich wird. Diese beiden Kinetiken arbeiten jedoch mit unterschiedlichen
Zeitfaktoren, so dass eine Skalierung der jeweiligen Ratenmatrizen nötig ist, um die
resultierende Ratenmatrix zu verwenden.
Die Arbeit untersucht Möglichkeiten der Hybridisierung beider Kinetiken, und
zeigt grundsätzliche Limitierungen des Kinetikansatzes auf. Zudem wird eine Metrik für
den Vergleich von Kinetiken vorgestellt, um optische Unterschiede im Faltungsverhalten zu
quantifizieren. Dieses Mass wird schliesslich verwendet, um die Qualität der Hybridkinetik
zu bewerten.

Im Rahmen des Projektes soll eine Methode umgesetzt werden, um den folding
funnel von Modellmolekuelen mit diskreten Energielandschaften zu schaetzen. Der
zu erstellende Ansatz baut direkt auf vorangegangenen Arbeiten auf und
ergaenzt diese um neue Datenstrukturen und Methoden.
Die Implementierung soll auf der C++ Programmierbibliothek
Energy
Landscape Library (ELL) aufbauen und diese ggf. ergaenzen. Zudem soll ein
standalone Programm entwickelt werden, mit dem direkt folding funnel Studien
ermoeglicht werden. Fuer einige gegebene, vorhandene Modelle sollen hierzu die
gesampelten Daten mit bestehenden exakten Studien verglichen werden, um
Qualitaet und Laufzeit des neuen Ansatzes zu bestimmen..
F�r Hintergrunddetails siehe pdf-Version der Projektbeschreibung.
Protein structure prediction has been always a very interesting problem to solve,
especially in the last 10 years. Many previous methods tried to focus on HP model
prediction, however most of those methods have the drawback of giving approximate
solutions. Another drawback of most of those works is that they ignore the
representation of the side chains of the amino acids.
In this master thesis, we develop an approach of a concrete constraint model
that takes the side chains of the amino acids into consideration and gives the exact
solutions for a given sequence in the side chain HP model in terms of minimizing the
energy, without any approximation.
In this work we also present the obtained results of the predication model and
show some important statistics especially about the degeneracy. In addition to that
we present some interesting results on generating protein like sequences, although this
is a hard task in the model. The protein like sequences can be found in a very low
probability in random sequences in the side chain model as we explain in this thesis.

Im Projekt soll ein Constraint-Programm zur Konstruktion kompakter Punktmengen im kubischen Gitter implementiert werden (Kernkonstruktion). Es soll dabei die C++-Bibliothek Gecode zur Anwendung kommen. Das Problem ist ein wesentlicher Baustein der constraint-basierten Proteinstrukturvorhersage in vereinfachten Gittermodellen.
Für das kombinatorische Problem der Kernkonstruktion existieren bereits ausführliche theoretische Vorarbeiten, sowie eine Implementierung in Mozart/Oz. Es geht daher ausdrücklich um eine effiziente Reimplementierung. C++- oder Javakenntnisse, sowie Erfahrung mit Constraintprogrammierung, bzw. die Bereitschaft zur tieferen Einarbeitung in diese Technik sind für das Projekt vorausgesetzt.
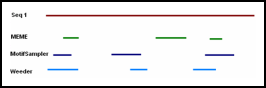
The project tackled the question : Can we trust the results of tools for de novo motif (TFBS) detection? If not, how can we improve the results?
Das Projekt beschäftigt sich mit Faltungsprozessen von Proteinstrukturen.
Im besonderen werden vereinfachte Gitterproteine betrachtet, um die
Komplexität der wirkenden Kräfte einzuschränken. Das
Konzept diskreter Energielandschaften bildet die Grundlage der Studie
und liefert das notwendige Framework.
Um Faltungsprozesse zu beschreiben müssen geeignete Umwandlungsoperationen
auf Proteinstrukturen definiert werden. Eine Möglichkeit ist die
Anwendung sogenannter Pull Moves, die lokale Änderungen der Struktur
bewirken.
![]() Im Rahmen des Projektes sollen die Pull Move Operationen in die generische C++
Programmierbibliothek
Energy Landscape Library (ELL)
integriert werden.
Darauf basierend soll anschliessend ein Standalone Programm entwickelt
werden, welches für eine gegebene Sequenz Faltungssimulationen
ermöglicht. Für einen gegebenen Satz von Testsequenzen soll
anschliessend mit Hilfe des Tools eine Klassifikation in 'gut' und
'schlecht' faltende Sequenzen erreicht werden. Hierzu müssen passende
Parameter und Kriterien gefunden werden.
Im Rahmen des Projektes sollen die Pull Move Operationen in die generische C++
Programmierbibliothek
Energy Landscape Library (ELL)
integriert werden.
Darauf basierend soll anschliessend ein Standalone Programm entwickelt
werden, welches für eine gegebene Sequenz Faltungssimulationen
ermöglicht. Für einen gegebenen Satz von Testsequenzen soll
anschliessend mit Hilfe des Tools eine Klassifikation in 'gut' und
'schlecht' faltende Sequenzen erreicht werden. Hierzu müssen passende
Parameter und Kriterien gefunden werden.
In this thesis we have developed two pairwise comparison methods on the basis of exact matching substructures, called exact pattern matches. In a first step, a set of overlapping and crossing substructures for two nested RNA secondary structures is found with the approach of pairwise common substructures from Siebert/Backofen. Our first method deals with the task to identify the best global subset of Non-Crossing exact pattern matches for two given RNAs. In relation to the LAPCS problem, we call this problem the Longest Common Subsequence of Exact RNA Patterns. The developed dynamic programming algorithm needs O(n�m�) time and O(nm) space. Our second approach detects (local) clusters of exact pattern matches. A cluster is a Non-Crossing arrangement of exact pattern matches with a distance constraint between the substructures included in a cluster. The developed clustering strategy to find clusters is fast and flexible enough for different analytical problems. We have tested both methods with two Hepatitis C virus RNAs and two 16s ribosomal RNAs. The results show that both methods are able to identify significant similarities between two RNA secondary structures in a fast way.
Genetic Regulatory Networks are a method of representing the complex assemblages of interconnected genes, proteins and other molecules. Components are represented as nodes, and activations and repressions between them are represented as labeled edges. Within these networks are Key Regulators; these are nodes capable of regulating a sub-network of the network. The task at hand is to present a model for genetic regulatory networks and to implement a means to identify the most suitable key regulator within the network. In addition, cycles representing positive or negative feedback loops increase the complexity of the task. The model is further refined by considering constraints such as choosing a key regulator that regulates a certain sub-network but has no effect on another sub-network.
Comparison of RNAs is mainly based on information about the sequence and their secondary structure. The function of RNAs on the other hand is based on their 3D-Structure, which is hard to determine. However, there are wide-spread 3D motifs which can be identified more easily. Such a motif can be defined, due to Eric Westhof, as an ordered assembly of non-watson-crick basepairs within a helix. Current sequence structure alignment methods are not aware of such motifs. However, these motifs can give strong guidance for such alignments. The project's aim is to integrate the knowledge about motifs into the recent tool LocARNA, which is a program for simultaneous folding and alignment of RNAs. The dynamic programming algorithm should be modified to detect the motifs and tested on biological data.
The structures of RNA molecules and proteins, which are both important biopolymers,
are commonly assumed to be uniquely determined by their sequences. The structures
of these biomolecules are in turn necessary to carry out the molecules' biological functions.
Discretized structure models provide a coarse-grained description of the molecular
structure, which is necessary to perform computational studies. In this research, RNA
molecules were modeled as secondary structures for RNA, and proteins were modeled as
self-avoiding walks on a lattice. The structure formation process of biopolymers is crucially
determined by the properties and the topology of the underlying energy landscape,
in which the folding proceeds. Typical characteristics of the energy landscape, like the
number of local optima, the basin distribution as well as the transition states between the
optima, can be visualized by barrier trees. Barrier trees provide a reduced representation
of energy landscapes, which can be used to study the dynamical behavior of biopolymer
folding.
The research described in this thesis aimed to present a generic, problem-independent
approach for the generation of barrier trees.
Many problems and open questions of modern microbiology demand the comparison of RNA, DNA or protein sequences. The computational effort in performing these calculations is high and microbiology urges the development of efficient tools. An important requirement is the capability of these tools to deal with certain constraints. Examples might be a specified number of matches of two compared sequences within certain regions or a consideration of the secondary structure of a proteine. Subject of this thesis is the efficient alignment of sequences, where we focus especially on different constraints on the alignment and how to combine these constraints, even in multiple alignments.
The standard depth first search (DFS) method to solve Constraint Satisfaction Problems (CPSs) shows much redundant work if the CSP contains several unsolved independent partial problems. This is a frequent observation when constraint programming is applied to solve the structure prediction problem in the HP model. Within this thesis, an implementation of a dynamically decomposing search strategy is developed and implemented. The resulting prototypical implementation is applied to the structure prediction problem and yields significant speedups compared to standard DFS. This speedup is neccessary e.g. to allow for high throughput degeneracy calculation of lattice proteins in the HP model.